Importing the literature network
Find all Python code used on this page here: metalit_setup.py
Before we can upload the literature network to Neo4j, we first need to create the appropriate files to upload. Specifically, we need to convert the XML file to a metadata file that can be imported easily. We also need to make sure the taxon identifiers match. The NJS16 network only has species identifiers, not the full taxonomy, so we will take the taxonomy of nodes in the association network and match this to the NJS16 identifiers.
First load the necessary packages and files through a Python interpreter. Make sure to set the loc parameter to the location where you stored the files. For instructions on starting a Python interpreter, please see the API section of the manual.
import networkx as nx
from mako.scripts.io import IoDriver
import os
import xml.etree.ElementTree as et
import pandas as pd
loc = os.getcwd()
Next, we will use the xml package to parse the NJS16 file. We are going to simplify interaction names a bit, to make later queries a bit simpler. Finally, we will construct a list of dictionaries that contains all the information we need to upload to Neo4j.
xtree = et.parse(loc + "/NJS16_MetabolicActivity_as_EdgeAttribute.xml")
xroot = xtree.getroot()
node_dict = {}
edge_rows = []
# Simplify interaction types for Neo4j relationship use
interaction_names = {'Consumption (import)': 'consumption',
'Consumption (import) & production (export)': 'consumption_production',
'Macromolecule degradation': 'macromolecule_degradation',
'Production (export)': 'production'}
# Make a dictionary linking node ID to node label
for node in xroot:
if node.tag == '{http://www.cs.rpi.edu/XGMML}node':
node_dict[node.attrib.get("id")] = node.attrib.get("label")
# Make for each microbe-metabolite interaction, a dictionary containing source, target and interaction
for node in xroot:
if node.tag == '{http://www.cs.rpi.edu/XGMML}edge':
children = list(node)
interaction = None
for child in children:
if child.attrib.get("name") == 'interaction':
interaction = child.attrib.get("value")
edge_rows.append({"source": node_dict[node.attrib.get("target")],
"target": node_dict[node.attrib.get("source")],
"interaction": interaction_names[interaction]})
There is one peculiarity we need to deal with: the include_nodes function that we are going to use, does not add metadata for nodes that do not currently exist in the database. Therefore, we will first run a custom query that creates all source nodes as Microbe nodes. This is also necessary because not all metabolite consumers / producers in the microbe/metabolite are actually microbes; colonocytes, for example, will not show up in our association network derived from metabarcoding data.
We first start the driver and run a batch query to address this. The edge_rows list is already in a format that can be used for these queries. The driver will return all names of the nodes it created, we can simply ignore those. Next, we will add the remainder of the list of dictionaries using the include_nodes function. This function uses the name and label parameters to identify node labels, while the nodes parameter should be a list of dictionaries containing source and target nodes.
driver = IoDriver(uri='neo4j://localhost:7688',
user='neo4j',
password='test',
filepath=loc,
encrypted=False)
query = "WITH $batch as batch " \
"UNWIND batch as record " \
"MERGE (a:Microbe {name:record.source}) RETURN a"
driver.write(query, batch=edge_rows)
driver.include_nodes(nodes=edge_rows, name="Metabolite", label="Microbe")
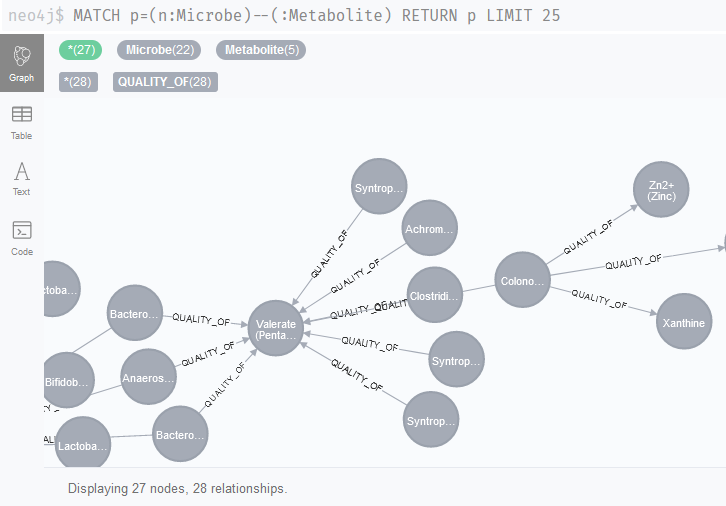
Now that the entire literature network has been uploaded to Neo4j, we can start querying this network through Neo4j (Figure 1), for example with the query MATCH p=(n:Microbe)--(:Metabolite) RETURN p LIMIT 25
However, there is one major issue we need to solve: the Sung et al. network has complete species identifiers, but our association network does not. Therefore, we will parse the Sung et al. nodes to connect them to Genus identifiers; since nearly all of our association networks do have those connections, we should be able to link our association network to metabolites that way. We can similarly use the include_nodes function to connect our nodes.
genus_dict = []
for edge in edge_rows:
microbe = edge['source']
genus_dict.append({'source': microbe,
'target': 'g__' + microbe.split(' ')[0]})
driver.include_nodes(nodes=genus_dict, name="Genus", label="Microbe")
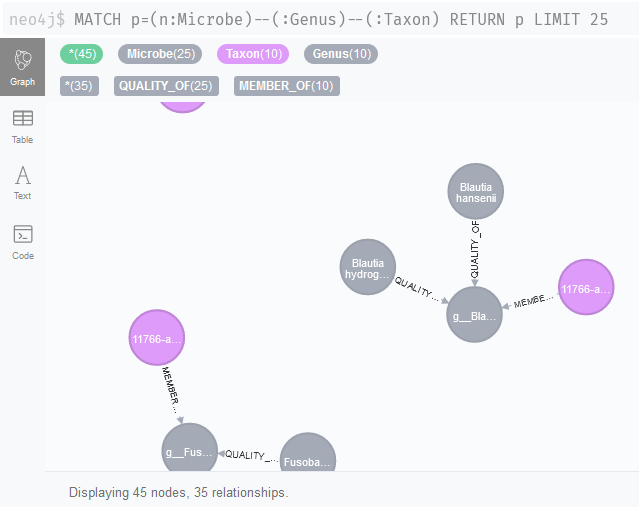
Our association network nodes are stored as Taxon nodes. Can we link these nodes to the literature network now (Figure 2)? Indeed, it looks like we should be able to identify associations that could be linked to specific metabolites with queries like MATCH p=(n:Microbe)--(:Genus)--(:Taxon) RETURN p LIMIT 25