Finding HMA-LMA subgraphs
Find all Python code used on this page here: sponge_queries.py
With all networks and the HMA-LMA links available via the Neo4j database, we can start considering associations that could be linked to HMA-LMA status. For example, we might try to recover all associations between taxa that are both associated to HMA status in some way, with the query below:
MATCH p=(:Type {name: 'HMA'})--()--(:Taxon)--(:Edge)--(:Taxon)--()--(:Type {name: 'HMA'}) RETURN p LIMIT 25
This query first matches the HMA node and then passes through either a Class or Phylum node to connect to an association, which should similarly be connected to the HMA node.
Cytoscape is an open source platform for visualizing networks; if you have not yet installed it, download it from the Cytoscape homepage. If we want to export the subgraph of HMA and LMA associations to Cytoscape, we can adapt the above query to connect associations to a new network node. The easiest way to do this, is via the mako API, which you can access via a Python interpreter. For instructions on starting the interpreter, please see the API section of the manual. Make sure to adjust the driver configuration settings for your instance of Neo4j.
from mako.scripts.io import IoDriver
import os
loc = os.getcwd()
driver = IoDriver(uri='neo4j://localhost:7688',
user='neo4j',
password='test',
filepath=loc,
encrypted=False)
driver.write("MERGE (n:Network {name: 'HMA_network'}) RETURN n")
hma_query = "MATCH p=(:Type {name: 'HMA'})--()--" \
"(:Taxon)--(a:Edge)--(:Taxon)--()--(:Type {name: 'HMA'}) RETURN a"
results = driver.query(hma_query)
First, the driver is used to create a HMA Network node. The query after that returns all Edgenodes that are linked to HMA status in these sponges. The query below takes the names of these nodes and connects them to the HMA node.
edge_names = [{"name": x['a']['name']} for x in results]
query = "WITH $batch as batch " \
"UNWIND batch as record " \
"MATCH (a:Edge {name:record.name}), (b:Network {name: 'HMA_network'})" \
"MERGE (a)-[r:PART_OF]-(b) RETURN r"
driver.write(query, batch=edge_names)
We can repeat the above queries for the LMA status.
driver.write("MERGE (n:Network {name: 'LMA_network'}) RETURN n")
lma_query = "MATCH p=(:Type {name: 'LMA'})--()--" \
"(:Taxon)--(a:Edge)--(:Taxon)--()--(:Type {name: 'LMA'}) RETURN a"
results = driver.query(lma_query)
edge_names = [{"name": x['a']['name']} for x in results]
query = "WITH $batch as batch " \
"UNWIND batch as record " \
"MATCH (a:Edge {name:record.name}), (b:Network {name: 'LMA_network'})" \
"MERGE (a)-[r:PART_OF]-(b) RETURN r"
driver.write(query, batch=edge_names)
Finally, we can export these two networks to a running instance of Cytoscape.
driver.export_cyto(networks=['HMA_network', 'LMA_network'])
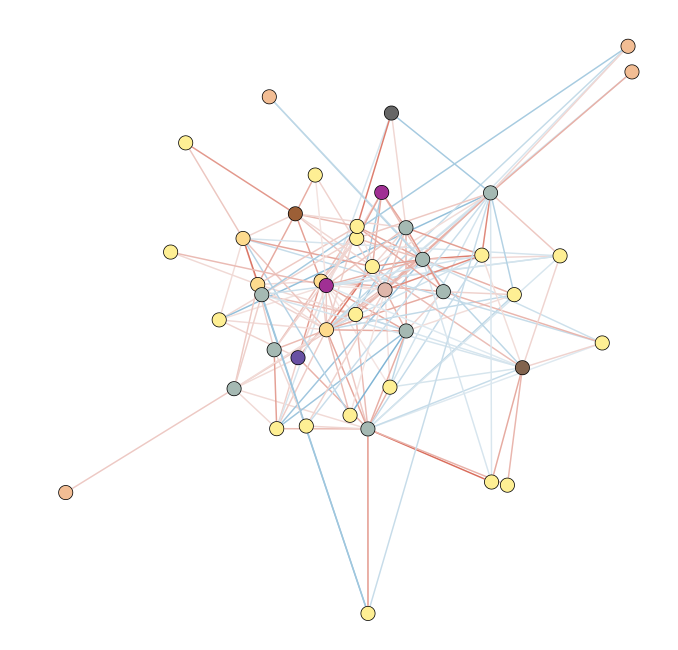
With Cytoscape, it is possible to format networks based on specific properties (Figure 2). Indeed, it becomes possible to see that most associations in the LMA network are Gammaproteobacteria (shown in yellow). Additionally, the mako export includes information like the number of source networks that contained these associations; although not shown below, it looks like most associations between taxa linked to LMA status only occur in one of the sponge networks, rather than in multiple of them.